
Topogivity: A Machine-Learned Chemical Rule for Discovering Topological Materials
Authors
Andrew Ma, Yang Zhang, Thomas Christensen, Hoi Chun Po, Li Jing, Liang Fu, Marin Soljačić
Abstract
Topological materials present unconventional electronic properties that make them attractive for both basic science and next-generation technological applications. The majority of currently known topological materials have been discovered using methods that involve symmetry-based analysis of the quantum wavefunction. Here we use machine learning to develop a simple-to-use heuristic chemical rule that diagnoses with a high accuracy whether a material is topological using only its chemical formula. This heuristic rule is based on a notion that we term topogivity, a machine-learned numerical value for each element that loosely captures its tendency to form topological materials. We next implement a high-throughput procedure for discovering topological materials based on the heuristic topogivity-rule prediction followed by ab initio validation. This way, we discover new topological materials that are not diagnosable using symmetry indicators, including several that may be promising for experimental observation.
Concepts
The Big Picture
Imagine identifying every poisonous plant on Earth not by testing each one in a lab, but by glancing at the leaf shape and guessing. Reckless for botany. But researchers at MIT just did something similar for topological materials: they found a rule simple enough to compute by hand that predicts, with roughly 80% accuracy, whether a material harbors exotic quantum properties. The only input is its chemical formula.
Topological materials are an unusual class of matter whose electrons are governed by topology, the branch of geometry that distinguishes a donut from a sphere. If you know the topological shape, you know which behaviors are possible, no matter how you deform the material without tearing it.
The practical consequences are real: electrical currents that flow along surfaces and shrug off defects, electrons that act like massless particles, phenomena relevant to quantum computers and next-generation electronics.
Finding these materials has been slow going. The standard approach analyzes quantum wavefunctions (mathematical descriptions of electron behavior) using a crystal’s geometric symmetry. It requires serious computing power, and it fails entirely for materials whose atomic arrangements lack the right kind of symmetry. A whole class of candidates stays hidden.
A team led by physicists Andrew Ma, Yang Zhang, and Marin Soljačić took a different approach. Using machine learning, they derived a single number for each chemical element, a quantity they call topogivity, and showed that a simple weighted average of these numbers predicts whether a compound is topological.
Key Insight: Topogivity assigns each element a learned score capturing its tendency to form topological compounds; the weighted average of a material’s elements’ topogivities reveals whether that material is likely topological or trivial, no quantum calculation required.
How It Works
The training data comes from existing first-principles databases: collections of materials whose quantum-mechanical properties have been computed from scratch. It covers non-magnetic, three-dimensional compounds with fixed elemental ratios, each classified as either “topological” or “trivial.”
The researchers deliberately chose an interpretable ML model, not a deep neural network but something far simpler. Their goal wasn’t just prediction accuracy. They wanted insight.
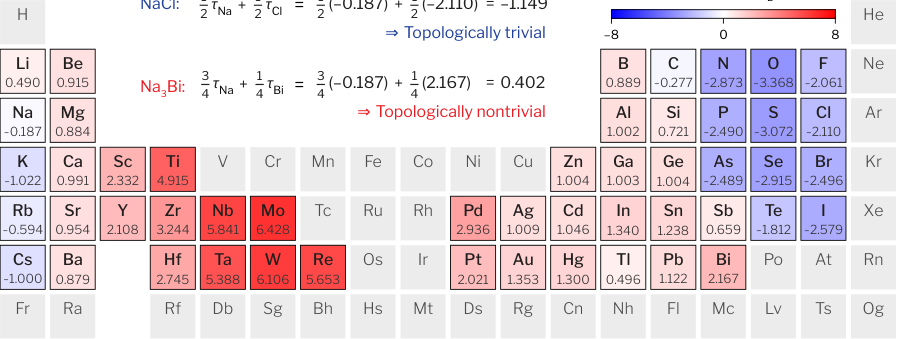
The heuristic rule works like this:
- Assign a topogivity value (τ) to each element in the periodic table, a single machine-learned number encoding how strongly that element tends to appear in topological compounds.
- Compute the weighted average of the constituent elements’ topogivities, weighted by their stoichiometric ratios (the proportions in which each element appears in the formula). For compound AB: ½τ_A + ½τ_B. For AC₃: ¼τ_A + ¾τ_C.
- Read the sign: a positive result predicts topologically nontrivial; negative predicts trivial. Magnitude gives a rough confidence estimate.
This napkin-computable rule hits roughly 80% classification accuracy, even though it ignores crystal structure, symmetry, and electronic wavefunctions entirely.
What the ML model captured is a chemical intuition: certain elements, especially heavy atoms with large spin-orbit coupling (a quantum interaction between an electron’s spin and its orbital motion that grows stronger in heavier elements), tend to promote topological behavior. Others suppress it. Topogivity makes that intuition quantitative.
Where this really earns its keep is in what it can see that conventional methods cannot. Symmetry indicators, the dominant computational tool for detecting topological character, work by finding mathematical patterns in electron behavior that mirror a crystal’s geometric symmetry. They miss materials with low crystal symmetry or topological properties that don’t couple to symmetry at all. That includes Chern insulators and time-reversal-invariant Z₂ topological insulators, two classes defined by topological invariants that symmetry analysis cannot detect. The first experimentally confirmed Weyl semimetal, tantalum arsenide (TaAs), falls into this blind spot. Topogivity does not.
The team built a high-throughput discovery pipeline around this idea. First, screen a large candidate space with the fast topogivity rule. Then validate survivors with expensive density functional theory (DFT) calculations, a standard method that computes electronic structure from quantum-mechanical first principles. This two-stage funnel reserves DFT for promising candidates only, cutting computational cost by a wide margin. The pipeline turned up new topological materials that standard database searches would have missed.
Why It Matters
Most ML tools in materials discovery involve deep networks that are accurate but opaque. They find patterns without explaining them. By insisting on interpretability, the MIT team extracted a principle rather than just a prediction engine.
The analogy to electronegativity is deliberate. Electronegativity is itself a heuristic, not a rigorous quantum observable, yet it shapes much of chemical reasoning. Topogivity targets the same conceptual niche: a quick gut-check before committing to heavier calculation.
It’s also a testable hypothesis about the chemical origins of electronic topology. As ML becomes standard in materials discovery, what models understand matters as much as what they predict. A machine-learned number that slots into the same framework as electronegativity makes the case concretely: AI can quantify chemical intuition in regimes where human intuition runs out.
Open questions remain. Topogivity operates only on stoichiometry and ignores crystal structure, which limits its precision. Future extensions might incorporate structural motifs or learn separate topogivities conditioned on bonding environment. Whether analogous heuristics exist for magnetic topological materials, which the current framework excludes, is still an open problem.
Bottom Line: By distilling the quantum complexity of electronic topology into a single number per element, topogivity makes materials discovery faster and broader, catching topological materials that standard symmetry-based methods cannot see.
IAIFI Research Highlights
Machine learning can extract human-interpretable chemical rules from quantum-mechanical databases, connecting AI pattern recognition with the physics of electronic band topology.
Prioritizing interpretable over black-box ML models can yield transferable scientific principles, showing how AI can generate insight, not just predictions.
Topogivity provides the first broadly applicable chemical heuristic for identifying topological insulators and semimetals, including non-symmetry-diagnosable materials inaccessible to existing symmetry-based approaches.
Future work could extend topogivity to magnetic materials and structure-conditioned variants; full methodology and discovered candidates are detailed in [arXiv:2202.05255](https://arxiv.org/abs/2202.05255).
Original Paper Details
Topogivity: A Machine-Learned Chemical Rule for Discovering Topological Materials
2202.05255
["Andrew Ma", "Yang Zhang", "Thomas Christensen", "Hoi Chun Po", "Li Jing", "Liang Fu", "Marin Soljačić"]
Topological materials present unconventional electronic properties that make them attractive for both basic science and next-generation technological applications. The majority of currently known topological materials have been discovered using methods that involve symmetry-based analysis of the quantum wavefunction. Here we use machine learning to develop a simple-to-use heuristic chemical rule that diagnoses with a high accuracy whether a material is topological using only its chemical formula. This heuristic rule is based on a notion that we term topogivity, a machine-learned numerical value for each element that loosely captures its tendency to form topological materials. We next implement a high-throughput procedure for discovering topological materials based on the heuristic topogivity-rule prediction followed by ab initio validation. This way, we discover new topological materials that are not diagnosable using symmetry indicators, including several that may be promising for experimental observation.